Control and Execution of Mitosis
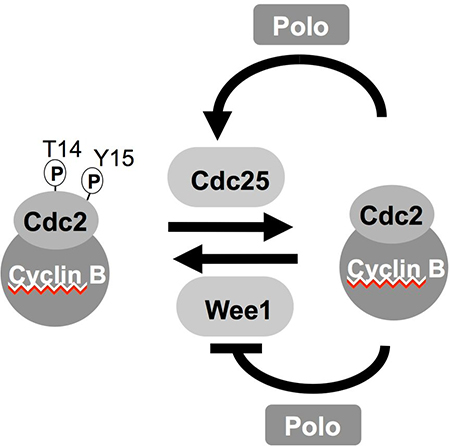
Commitment to mitosis is triggered by activation of the Cdk1-Cyclin B protein kinase complex. During interphase Wee1 related kinases phosphorylate the catalytic, Cdk1 subunit to inhibit the complex. This phosphate is removed by Cdc25 phosphatases to promote mitotic entry. Cdk1-Cyclin B activation promotes a positive feedback loop that boosts Cdc25 and inhibits Wee1 activities to ensure that mitotic commitment is a rapid and irreversible bi-stable switch. In many systems this feedback loop includes polo kinase. Although Cdk1-Cyclin B directly phosphorylates a limited number of molecules that mediate chromosome segregation to modify their behaviour, the principle mechanism by which Cdk1-Cyclin B promotes mitosis is by activating a set of downstream kinases that includes kinases of the Polo, Aurora, NimA kinases alongside the kinases Mps1, Haspin, Casein Kinase 1 and Bub1.
Our studies of mitotic commitment have gravitated towards the role played by the spindle pole in determining the timing of entrance to and exit from mitosis (see link on left for further details).
Our studies of mitotic execution kinases have focused upon the fission yeast polo, aurora and NIMA related kinases (Plo1, Ark1 and Fin1 respectively). We have employed a variety of approaches, ranging from localisation, activity assays, generation of mutant alleles to place the controls exerted by these kinases into the context of mitotic progression. The isolation of molecules under denaturing conditions has enabled us to map sites of phosphorylation by quantitative mass spectrometry. We use information about how the level of phosphorylation at a particular site changes in response to changes in cell cycle status or other contexts, alongside the degree of conservation of the amino acid in equivalent kinase of other species to guide the generation of mutant alleles. We introduce mutations to block or mimic phosphorylation at phosphorylated serine or threonine residues. Mutations that show phenotypes that are consistent with phosphomimetic or phosphoblocking behaviour are crossed into a variety genetic backgrounds to interrogate kinase function.
We have also applied the approach of genetically sensitizing protein kinases to inhibitory ATP analogues that was developed by Kevan Shokat and colleagues. In this approach mutations are into the gatekeeper residues on the edge of the ATP binding pocket. The increased size of the catalytic pocket enables bulky ATP analogues to access the pocket. As the only kinase into which such bulky analogues can fit is the mutated kinase, the analogue becomes specific, selective, inhibitor of the target kinase. For Plo1 and Wee1 kinases we identified a further mutation at the back of the ATP binding pocket that must be mutated in order to sensitize the kinase to analogue inhibition.
A final approach that has been particularly informative in our study Plo1 function has been a collaborative venture with Boris Maček’s lab in Tübingen in which we use SILAC labelling to identify sites where phosphorylation changes when polo kinase activity is elevated or depressed. The use of this phospho-proteomics data to guide mutagenesis of key phosphorylation sites is revealing Plo1 participation in some unanticipated areas of biology.