Controlling Entrance To and Exit From Mitosis from the Spindle Pole
Centrosomes nucleate the two microtubule arrays that inter-digitate to form the mitotic spindle. A number of observations, including the initial appearance of active Cdk1-Cyclin B on human centrosomes and modelling of Cdk1-Cyclin B activation in frog egg extracts, suggest that the trigger for mitotic commitment stems from the spindle pole. Our studies of a component of the fission yeast centrosome equivalent, the spindle pole body (SPB), strongly endorse this view.
In fission yeast, we find that Cdk1Cdc2-Cyclin B activity appears on the SPB 30 minutes before mitosis while the rest of the cell remains in an interphase state. Cdk1Cdc2-Cyclin B activity activates the feedback loop kinase PoloPlo1 that is recruited to the SPB as soon as the SPB bound Cdk1Cdc2-Cyclin B is activated. Genetic evidence strongly suggests that this activation on the spindle pole triggers the eventual commitment to mitosis. Gain of function mutations in the structural SPB component Cut12 (e.g. cut12-s11) compensate for an otherwise lethal loss of Cdc25 function in the temperature sensitive cdc25-22 at 36°C. This influence appears to stem from an influence over PoloPlo1activity as the cut12 mutations that suppress deficiencies in Cdc25 both advance the point in G2 phase at which PoloPlo1 is recruited to the SPB and boost PoloPlo1 kinase activity throughout the cell. This enhancement of PoloPlo1 activity would increase the inhibitory feedback loop activity towards Wee1. With Wee1 inhibited there would be less phosphate on Cdk1Cdc2 and so less or no requirement for Cdc25 phosphatase activity to remove phosphate from Cdk1Cdc2.
Triggering Mitotic Commitment from the SPB
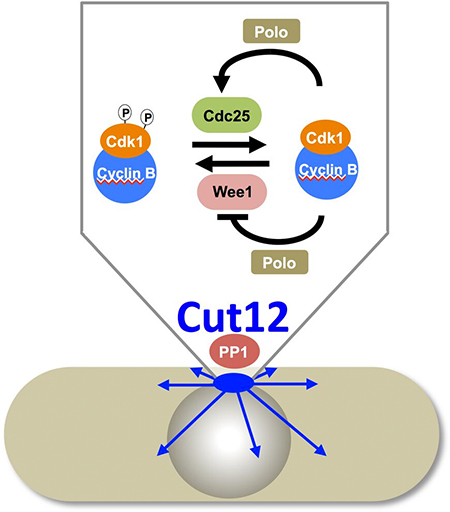
We have taken a direct approach to follow up this correlative evidence that centrosomes/SPBs play a key role in mitotic commitment by asking the simple question “Would Cdk1Cdc2-CyclinB/ PoloPlo1activation anywhere in the cell be sufficient to trigger mitosis, or is there something special about the SPB that would support mitotic commitment?” We fused each kinase to a single chain llama antibody called GFP binding protein (GBP). As GBP has a very high affinity for the Green Fluorescent Protein (GFP) we used GBP to direct PoloPlo1 and Cdk1Cdc2 kinases to a wide range of structures defined by a variety of GFP fusion proteins. Each kinase incorporated a mutation that rendered it constitutively active and insensitive to normal cell cycle controls. Their catalytic pockets were further modified to incorporate mutations that conferred sensitivity to inhibition by ATP analogues. We could then restrain the activities of the targeted kinases with inhibitory ATP analogues until analogue removal by a simple exchange of culture medium provided a burst of kinase activity at the subcellular location of interest. We used this approach to release each kinase activity at a variety of subcellular locations including nuclear pores, centromeres and the cell cortex. The only location at which the release of either kinase activity triggered mitosis was the SPB. Moreover, of the three SPB components tested, the drive into mitosis was greatest when the chimeric kinases had been recruited by Cut12-GFP.
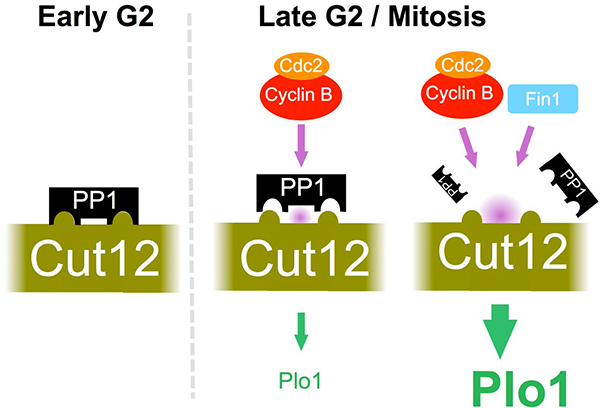
PP1 Recruitment to Cut12 Controls Mitotic Commitment
Protein phosphatase 1 (PP1) is recruited by defined docking motifs. The cut12 mutations that suppress cdc25-22 sit within a perfect match for a PP1 docking site. We employed a range of approaches to show that PP1 recruitment to this site in Cut12 sets the requirement for Cdc25 in fission yeast. Strikingly cells could tolerate the otherwise lethal ablation of the cdc25+ gene when the PP1 docking site had been removed from Cut12. We are yet to identify the target for the Cut12-PP1 complex however we did find an inverse correlation between the degree of PP1 recruitment to Cut12 and the SPB recruitment and activity of the feedback loop kinase PoloPlo1. Furthermore, PP1 recruitment to Cut12 constitutes yet another level of feedback control because PP1 docking was inhibited by phosphorylation of the docking site by Cdk1Cdc2-Cyclin B and the NIMA kinase Fin1. Thus, elevation of Cdk1Cdc2-Cyclin B kinase activity triggers a loop that reduces PP1 recruitment to Cut12 to boost PoloPlo1activity in order to further enhance Cdk1Cdc2-Cyclin B activity towards the PP1 docking site. This loop will greatly enhance the bi-stable nature of the mitotic commitment switch.
Co-ordinating Change at the SPB
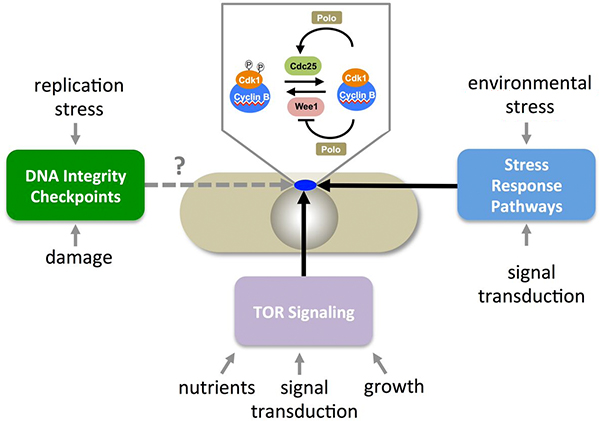
A screen to identify sites that maybe phosphorylated by aurora kinase led us to study the function of the serine at position 402 of PoloPlo1. Strikingly, we found that mutation the phosphomimetic plo1-S402E mutation suppressed cdc25-22 mutants, while the phosphoblocking mutation plo1-S402A blocked the ability of cut12-s11to suppress cdc25-22 deficiency. Furthermore, the premature recruitment of Plo1-GFP to the spindle pole of cut12-s11 mutants was abolished by plo1-S402A, whereas plo1-S402E enhanced recruitment to the SPB in otherwise wild type cells. Antibodies that only recognised PoloPlo1 when it was phosphorylated on S402 showed that phosphorylation of serine 402 of Polo Plo1 was under the control of the MAPKSty1 stress response pathway. Thus, phosphorylation of PoloPlo1 kinase to regulate its affinity for the SPB constitutes one mode of the previously enigmatic link between MAPKSty1 signalling and fission yeast mitotic control. As the strength of flux through the MAPKSty1 signalling cascade responds to changes in the nutrient environment, Janni Petersen, who did the PoloPlo1-S402 work, followed up her work on PoloPlo1-S402 in a senior post in Paul Nurse’s lab in Rockefeller University where she showed that PoloPlo1-S402 signalling changes radically when cells are shifted from rich to poor growth media. Changes in nutrient quality are sensed by the TOR signalling network. This TOR activity regulates Pyp2, one of the phosphatases that dephosphorylates MAPKSty1. In this way, changes in nutrient stress are sensed by the TOR network to change the activity of the MAPKSty1 kinase pathway towards S402 of PoloPlo1. Enhancement of PoloPlo1-S402 phosphorylation advances mitotic commitment, reduction in PoloPlo1-S402 signalling reduces it. Janni continues to study TOR sensing of nutrient status and its connection to mitotic control with her group in Adeleide.
The loss of DNA checkpoint responses in patients with a form of Seckels syndrome that arises from mutation of pericentrin indicate that the human centrosome plays a key role in co-ordinating the timing of mitotic commitment with DNA integrity. Whether such co-ordination is conserved in fission yeast and so is indicated by a grey dashed line in the representation above.
Plo1 Activation at SPBs Drives Growth at Cell Tips
MAPKSty1 signalling is stimulated by a variety of stresses. Strikingly, PoloPlo1-S402 phosphorylation plays a key role in regulating growth after centrifugation and heat stresses. Fission yeast growth is driven by actin dependent events at cell tips. Consequently F-actin patches accumulate at cell tips. When wild type cells are subjected to heat /centrifugation stress F-actin patches disassociate from cell tips before returning as growth resumes. The plo1-S402A mutation severely compromised the return of F-actin patches and consequent resumption of growth. Thus, something to do with phosphorylation is connected with morphogenesis at the cell tip. The subsequent analysis with the Plo1-GBP chimeric fusion proteins (described above) suggests that activation of PoloPlo1 on the SPB is the key event that drives tip growth at remote sites. Release of PoloPlo1 activity at cell tips was able to both enhance the density of F-actin at cell tips and activate dormant non-growing tips to convert them into actively growing tips.
Mitotic Exit and the SPB
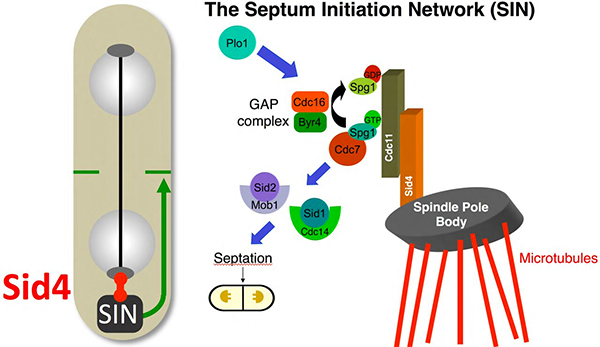
Fission of the S. pombe cell into two daughter cells is driven by the ordered formation and dissolution of a septum. Septation is driven by the activation of a network called the Septum Initiation Network (SIN). The SIN is the equivalent of Hippo signalling networks of higher eukaryotes. Like Hippo signalling, it assembles on a scaffold. The scaffold for the SIN is Cdc11. Conversion of a small GTPase, Spg1, into its GTP bound form recruits the PAK family kinase Cdc7. Cdc7 activation recruits then recruits the NDR/MOB kinase combination Sid2/Mob1. The Ste20 kinase Sid1 and its partner Cdc14 also associate with the complex and may couple SIN function to morphogenesis pathways at cell tips. SIN function relies upon anchorage of Cdc11 to the SPB via the SPB component Sid4. Weak SIN activity is associated with both SPBs upto the metaphase/anaphase transition, whereupon it disappears to return midway through anaphase B on the new SPB to drive septation.
The regulation of both mitotic commitment (via Cut12) and mitotic exit (via Sid4) on the SPB offers extensive opportunities for dialogue between entrance and exit controls to fine tune mitotic controls.