Overview
The inappropriate proliferation of cancer cells can arise from unchecked cell division, a failure to engage cell death pathways, or simultaneous changes in both. Understanding how the diverse cues are integrated to co-ordinate cell division and death is therefore key to understanding the biology of cancer. The DNA damaging approaches of chemotherapy and irradiation owe much of their success to the checkpoint pathways that ensure transition through the cell division cycle only occurs when genome integrity is guaranteed. We study these checkpoint pathways and the control of commitment to the physical process of genome segregation, mitosis. Because the regulatory networks that control cell division are highly conserved, we study these controls in human cells and unicellular fission yeast. The yeast work can identify key questions to ask in the context of complex human cell division cycle control.
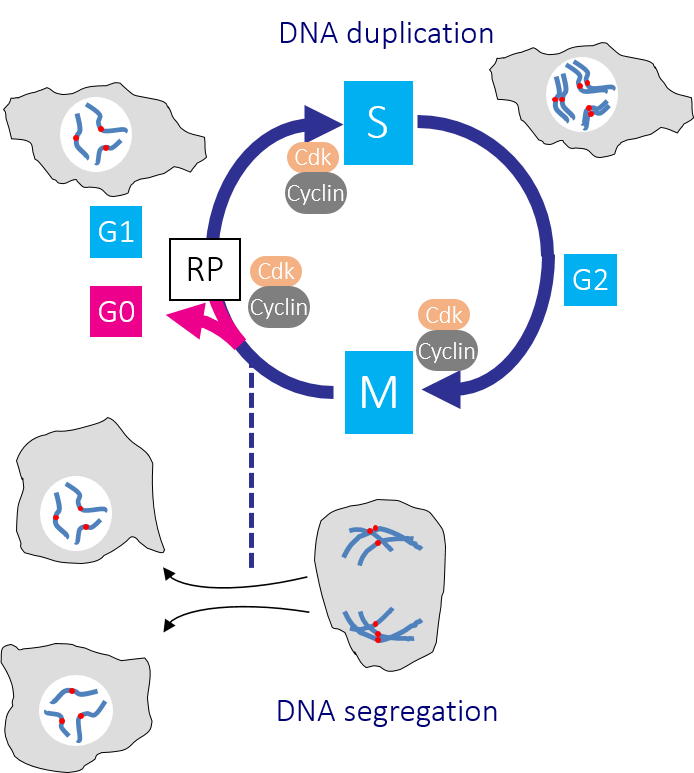 Figure 1: The human cell cycle with Cdk1-Cyclin B control of the G2/M transition Passage through the Restriction point (RP) in G1 phase commits a cell to passage through the cell division cycle. DNA replication in S phase is separated from mitosis by a gap phase, G2. Transition through the major rate limiting commitment steps into the cycle, replication and division is driven by Cdk-Cyclin activities.
Figure 1: The human cell cycle with Cdk1-Cyclin B control of the G2/M transition Passage through the Restriction point (RP) in G1 phase commits a cell to passage through the cell division cycle. DNA replication in S phase is separated from mitosis by a gap phase, G2. Transition through the major rate limiting commitment steps into the cycle, replication and division is driven by Cdk-Cyclin activities.
The cell cycle
In a typical cell division cycle the G1 gap phase precedes DNA replication in S phase, before a second gap phase G2, separates S from genome segregation in Mitosis (M phase) (Figure 1). Growth, developmental and environmental cues determine whether and when a cell leaves the non-cycling G0 state to enter the cell cycle by passing through a decision point of no return in G1 phase called the ‘Restriction point’ (Figure 1). Once cells are through the Restriction point, they are committed to the cycle and progress around the cycle, even if the pro-division cues that pushed them through this regulatory step are removed. Passage through the Restriction point and a second key regulatory step at the G2/M boundary is driven by the activation of distinct CDK-Cyclin protein kinase complexes. Successive waves of Cdk-cyclin activities drive different events as cells transit the cycle (Figure 2). The collective activity of all Cdk-Cyclin kinases must decline to zero in G1 phase because the recruitment of DNA replication proteins to origins of replication is inhibited by Cdk-Cyclin activity. If any Cdk-Cyclin activity persists in this crucial DNA replication licensing window, replication will be defective as some origins will fail to fire and others fire repeatedly.
Cancer and Cdk-Cyclin
In order to inappropriately drive cells through the restriction point and proliferate in the wrong circumstances, cancer cells boost Cdk-Cyclin. However, this de-regulated Cdk-Cyclin activity in the critical replication licensing window, when all Cdk-Cyclin activity should be repressed, causes DNA damage in a phenomenon that is known as ‘oncogene induced replicative stress’ (OIRS). OIRS can raise the degree of damage in cancer cells towards levels that are lethal. This is why the DNA damaging approaches of chemotherapy and radiotherapy are so effective in the clinic: the extra damage from therapy pushes the cancer cells over their lethality threshold.
A cancer’s need to repair more damage than neighbouring tissue places greater reliance upon DNA integrity cell cycle checkpoints that repress Cdk-Cyclin activities to delay cell cycle progression while the damage is repaired. Once the damage is repaired, checkpoint signalling abates and Cdk-Cyclin activity is restored, enabling passage through the cell cycle to re-start. As these pathways are so efficient at blocking division, cancers invariably inactivate the primary DNA damage checkpoint in which p53 blocks passage through the Restriction point. Such p53 deficient cancer cells become entirely reliant upon a second checkpoint at the G2/M boundary. There is therefore considerable therapeutic interest in weakening this second G2/M checkpoint as it will force cancer cells into a lethal division, while their normal neighbours can use their functional p53 checkpoint to pause the cycle when they are damaged.
G2/M transition
The G2/M transition is driven by activation of the Cdk1-Cyclin B protein kinase. Wee1 kinases inhibit Cdk1-Cyclin B by phosphorylating the catalytic Cdk1 subunit. Removal of this phosphate by Cdc25 phosphatases then promotes mitotic entry. A trigger level of Cdk1-Cyclin B activation promotes a positive feedback loop that employs Polo kinase to boost Cdc25 and inhibit Wee1 activities to ensure that mitotic commitment is a rapid and irreversible switch from one state (interphase) into another (division) (Figure 2).
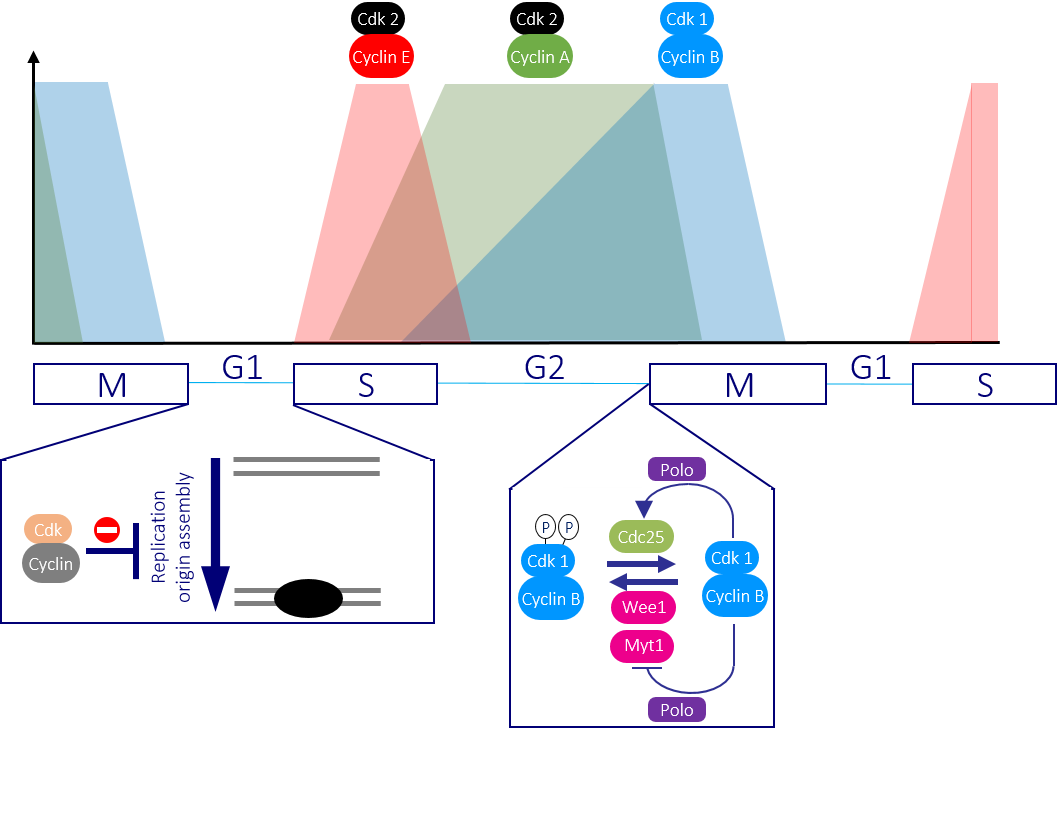 Figure 2: Successive waves of Cdk-Cyclin activity drive cells through the cycle Once cells are driven through the restriction point by Cdk4-Cyclin D and Cdk6-Cyclin D activities, Cdk2-Cyclin E and Cdk2-Cyclin A promote DNA replication. Cdk2-Cyclin A activity persists until the beginning of mitosis, which it triggers alongside the principle mitotic regulator Cdk1-Cyclin B. The absence of any Cdk-Cyclin activity is critical to ensure that the DNA replication complexes can assemble ready for the subsequent division.
Figure 2: Successive waves of Cdk-Cyclin activity drive cells through the cycle Once cells are driven through the restriction point by Cdk4-Cyclin D and Cdk6-Cyclin D activities, Cdk2-Cyclin E and Cdk2-Cyclin A promote DNA replication. Cdk2-Cyclin A activity persists until the beginning of mitosis, which it triggers alongside the principle mitotic regulator Cdk1-Cyclin B. The absence of any Cdk-Cyclin activity is critical to ensure that the DNA replication complexes can assemble ready for the subsequent division.
The checkpoint pathways that block mitotic commitment when DNA is damaged, or replication is incomplete, do so by boosting the activity of Wee1 family kinases and repressing Cdc25. Thus, when thinking about how to target the G2/M checkpoint, there has been considerable interest in developing drugs that inhibit Wee1. Most of this interest has focused upon the founder member of this kinase family, Wee1. However, while we know a lot about Wee1, we know remarkably little about the biology of the closely related Myt1 kinase. We are therefore investigating how, when and why Myt1 is used to block the commitment to mitosis. Our findings will guide the application of small molecule inhibitors that are currently under development.
Centrosomes and G2/M transition
A second major interest of the team is understanding the role played by the centrosome in regulating the G2/M transition. Centrosomes nucleate all the microtubules in the cell to generate the interphase cytoskeleton and the bipolar mitotic spindle that physically segregates the chromosomes. However, we believe that centrosomes organise more than microtubules. The initial appearance of active Cdk1-Cyclin B on human centrosomes, before propagation throughout the cell, suggests that this organelle provides a specific microenvironment to trigger the G2/M transition. Our studies of the fission yeast centrosome equivalent, the spindle pole body (SPB), provide molecular insight into how and why this switch may operate (Figure 3). We have shown that release of Cdk1-Cyclin B, or Polo kinase activity at the SPB, will drive cells into division.
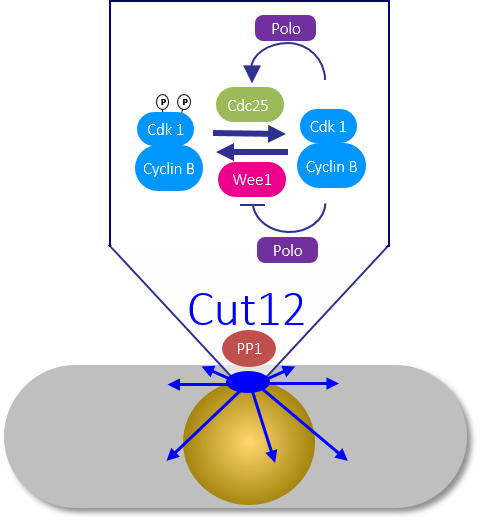 Figure 3: The fission yeast spindle pole body triggers mitotic commitment Recruitment of PP1 to the SPB component Cut12 determines the level of Polo kinase activity throughout the cell and so sets the threshold for the feedback loops that convert sparks of Cdk1-Cyclin B activity into a mitotic commitment wave that drives cells through division.
Figure 3: The fission yeast spindle pole body triggers mitotic commitment Recruitment of PP1 to the SPB component Cut12 determines the level of Polo kinase activity throughout the cell and so sets the threshold for the feedback loops that convert sparks of Cdk1-Cyclin B activity into a mitotic commitment wave that drives cells through division.
In contrast, release of either kinase activity at any other location around the cell has no impact upon division timing. Our attempts to define the molecular basis for such a striking impact have been guided by lessons from the SPB scaffold Cut12. Simply blocking the recruitment of protein phosphatase 1 (PP1) to Cut12 enabled us to delete the cdc25+ gene without compromising viability. This bypass of the requirement for an otherwise essential mitotic inducer arose from the impact of the Cut12/PP1 axis on Polo kinase activity. Polo activity was inappropriately elevated by the abolition of PP1 recruitment to Cut12. We are pursuing the hypothesis that Polo activity overcomes the need for Cdc25 because it boosts Polo’s ability to inhibit Wee1 to such a degree that it completely silences Wee1. In this scenario, the absence of the kinase that places the phosphate onto Cdk1 removes the need for the phosphatase that normally reverses the missing phosphorylation event.
By understanding how the trigger to initiate mitosis is flipped and how cell status can restrain this step, we will gather new insights in how to develop new therapies and optimise the exploitation of existing approaches to treatment options.