
Overview
Cancer is a genetic disease of inappropriate cell proliferation. Cell proliferation is a balance between increasing cell numbers, through cell division, and reducing them, through cell death. Within the cell division cycle, the duplication of the genome in S phase is separated from its segregation in mitosis. A complex structure called the mitotic spindle physically segregates the two genomes in mitosis. Pathways that promote cell death are initiated when cells enter mitosis with incomplete or damaged DNA, or when they become trapped in mitosis as a consequence of errors in chromosome architecture or spindle function. Because cancers have inherently high levels of genome instability, it is possible to find levels of DNA or spindle damaging agents that can trigger death in tumour cells, while leaving normal tissue untouched. Anti-mitotics and DNA damaging agents are therefore widely used in the clinic.
We exploit the simplicity and genetic malleability of yeast as a model system in which to identify fundamental principles in the control of the decisions to enter and exit mitosis. We define the molecular detail of conserved cell cycle controls and key principles of cell division in yeast for subsequent, highly focused, studies in much more complex human cell line models.
There are two principle themes to our research: the role played by the centrosome in promoting mitotic commitment and exit and the properties and control of the protein phosphatases that drive cells out of division. As cell division is accompanied by a cessation of migration and morphogenesis, some of our studies address very specific aspects of cytoskeletal control of morphogenesis.
Featured Publications
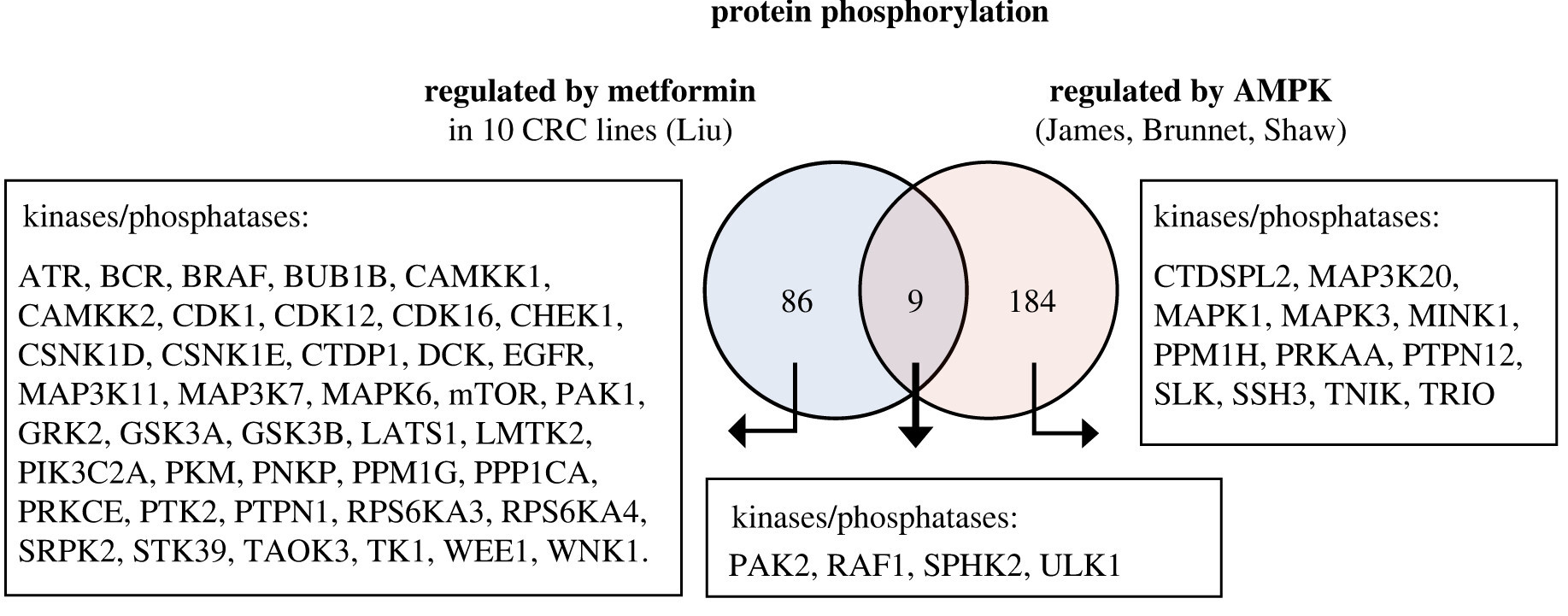
Elevated basal AMP-activated protein kinase activity sensitizes colorectal cancer cells to growth inhibition by metformin
12th April 2023
Authors find elevated basal AMP-activated protein kinase activity sensitises colorectal cancer cells to growth inhibition by metformin, with potential for repurposing an existing drug to treat cancer.

A TOR (target of rapamycin) and nutritional phosphoproteome of fission yeast reveals novel targets in networks conserved in humans
7th April 2021
Authors map TOR and nutrient-controlled signalling in fission yeast and identify nitrogen and TOR-regulated phosphorylation sites. Analysis provides source of potential substrates of TOR and N signalling that regulate diverse spectrum of biological processes.

Release from cell cycle arrest with Cdk4/6 inhibitors generates highly synchronized cell cycle progression in human cell culture
14th October 2020
Authors developed a new approach to synchronise the cell division cycle of an entire population of human cells in culture.
The cell cycle
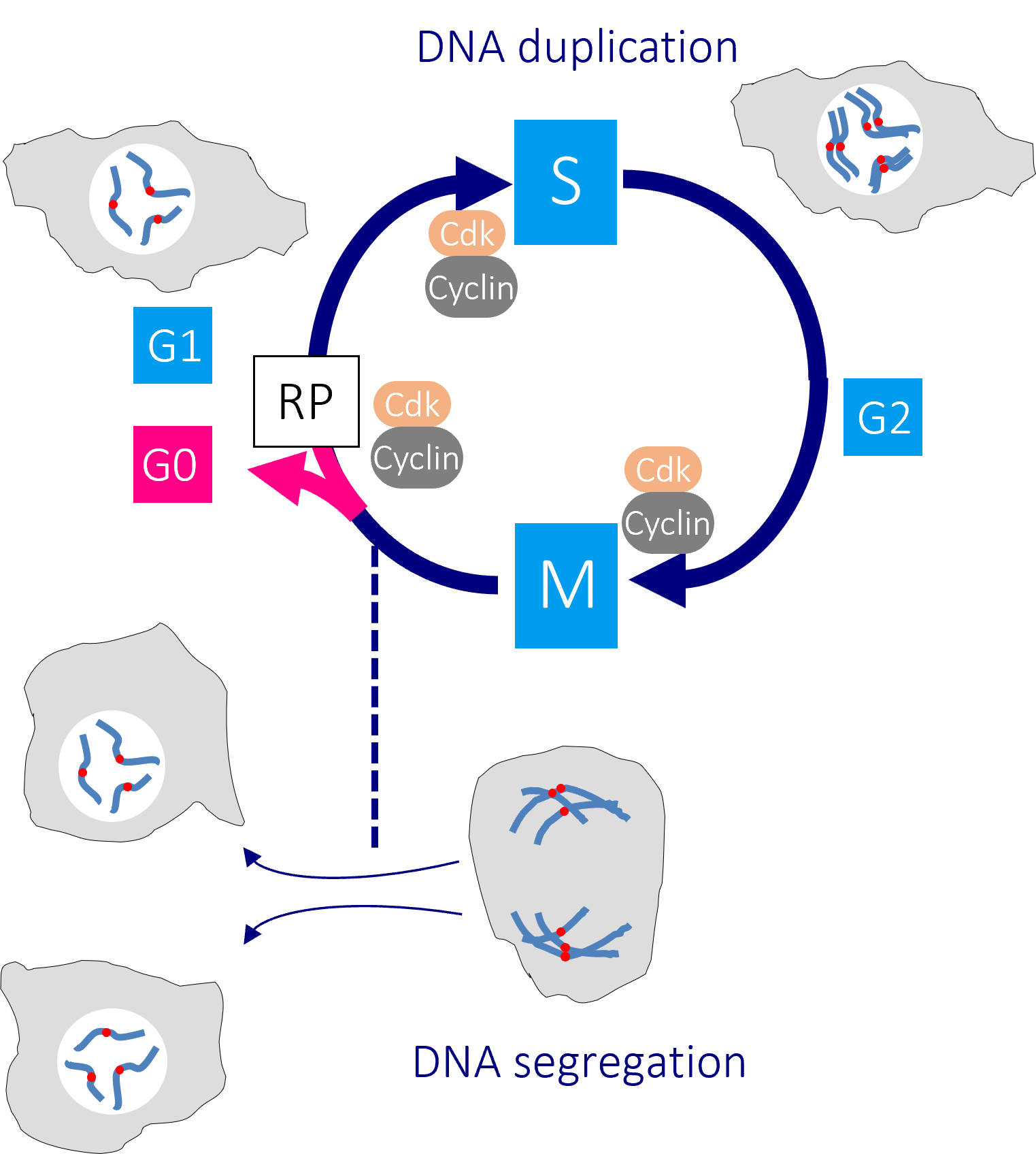
The human cell cycle with Cdk1-Cyclin B control of the G2/M transition
Passage through the restriction point (RP) in G1 phase commits a cell to passage through the cell division cycle. DNA replication in S phase is separated from mitosis by a gap phase, G2. Transition through the major rate limiting commitment steps into the cycle, DNA replication (S) and genome segregation (M) is driven by CDK-Cyclin activities.
Defects in DNA integrity activate cell cycle checkpoints that block progression through key cell cycle transitions until the damage is restored. As the mutations that enable cancer cells to bypass normal growth controls lead to the accumulation of DNA damage and changes to the chromosome number, cancer cells become more reliant upon these checkpoints than their normal neighbours.
Consequently, agents that enhance DNA damage are widely used in the clinic as they increase the level of damage in the already stressed cancer cells to a point where checkpoint defenses are unable to prevent catastrophic division. By contrast, their normal neighbours simply extend their cell cycle times to accommodate the elevated level of damage. We are therefore asking how these checkpoints operate to find ways to manipulate checkpoint controls in a manner that will selectively eliminate cancer cells.

Research areas
Meet the group
It’s a great pleasure to introduce the Cell Division group, who are a joy to work with as they are always eager to support each other and welcome new members to the team. They are dedicated to their research and the work we do in our lab. And I am delighted to say that they also find time to have fun as well!

Senior Group Leader

Postdoctoral Scientist
Senior Scientific Officer
Senior Scientific Officer

PhD Student

Senior Scientific Officer

Senior Scientific Officer

Senior Scientific Officer
Senior Scientific Officer

Sharing successes
Sharing successes
Colloquium 2024 Best Poster Prize winner
At the Institute’s annual Colloquium, the 2024 Best Poster Prize for a scientific officer went to Pawan Singh of the Cell Division group for his outstanding work on converting rising cdk1-cyclin b levels into a switch for mitotic commitment via feedback control from the fission yeast spindle pole body.
All Institute Publications
https://www.nature.com/articles/s41467-026-69964-2
Disruption of tRNA threonylation triggers RIG-I mediated anti-tumour immune response
25 February 2026
Institute Authors (1)
Sylvain Delaunay
Research Group
RNA Dynamics in Cancer
25 February 2026
https://doi.org/10.1038/s44161-025-00740-z
Single-cell profiling reveals three endothelial-to-hematopoietic transitions with divergent isoform expression landscapes
11 November 2025
Institute Authors (6)
Robert Sellers, John Weightman, Wolfgang Breitwieser, Natalia Moncaut, Michael Lie-a-ling, Georges Lacaud
Labs & Facilities
Computational Biology Support, Molecular Biology, Genome Editing and Mouse Models
Research Group
Stem Cell Biology
11 November 2025
https://doi.org/10.1136/jitc-2025-012527
Systemic immunosuppression from ultraviolet radiation exposure inhibits cancer immunotherapy
31 October 2025
Institute Authors (4)
Isabella Mataloni, Antonia Banyard, Garry Ashton, Amaya Virós
Labs & Facilities
Mass and Flow Cytometry, Histology
Research Group
Skin Cancer & Ageing
31 October 2025
https://aacrjournals.org/cancerdiscovery/article/doi/10.1158/2159-8290.CD-24-1224/766638/Glucocorticoids-Unleash-Immune-dependent-Melanoma
Glucocorticoids Unleash Immune-dependent Melanoma Control through Inhibition of the GARP/TGF β Axis
15 October 2025
Institute Authors (12)
Charles Earnshaw, Poppy Dunn, Shih-Chieh Chiang, Maria Koufaki, Massimo Russo, Kimberley Hockenhull, Erin Richardson, Anna Pidoux, Alex Baker, Richard Reeves, Robert Sellers, Sudhakar Sahoo
Labs & Facilities
Computational Biology Support, Visualisation, Irradiation and Analysis
Research Group
Cancer Inflammation and Immunity
15 October 2025
/wp-content/uploads/2025/09/Annual_Report_2024.pdf
2024 Annual Report
23 September 2025
23 September 2025
https://doi.org/10.1182/blood.2024028033
An in vivo barcoded CRISPR-Cas9 screen identifies Ncoa4-mediated ferritinophagy as a dependence in Tet2-deficient hematopoiesis
4 September 2025
Institute Authors (1)
Justin Loke
Research Group
Myeloid Cancer Biology
4 September 2025
Get in touch


Institute life in Manchester
We strive to make our community a welcoming, caring and enthusiastic one, fuelling ambition with opportunities for training and mentoring to help us all achieve our personal and professional goals.

“We are so pleased to have received the funding to enable us to test our hypothesis in the lab. If we can create a new medicine that can precisely target a specific type of cell within the tumour, and restore anti-cancer immune responses, this will be a game-changer for oesophageal cancer patients “
Sara Valpione
Former Institute Clinical Fellow and now Clinician in Residence within the CRUK National Biomarker Centre

“My charity bake sales – known as “David’s Great British Bake Off” – are always a hit, home baked products taste so much better than shop bought and are greatly appreciated by staff!”
David Jenkins
Purchasing Officer

“We’ve seen some remarkable responses, with an improvement for some patients within days. This is an early phase trial so there’s a lot more work to do. But the data we have so far is very encouraging and could help many thousands of people in the future”
Tim Somervaille
Senior Group Leader

“It is a pleasure to introduce my team who work to deliver our research goals. We work in a friendly and collaborative environment, supporting each other’s projects. “
Amaya Virós
CRUK Advanced Clinician Scientist Fellow
“We are so pleased to have received the funding to enable us to test our hypothesis in the lab. If we can create a new medicine that can precisely target a specific type of cell within the tumour, and restore anti-cancer immune responses, this will be a game-changer for oesophageal cancer patients “
Sara Valpione
Former Institute Clinical Fellow and now Clinician in Residence within the CRUK National Biomarker Centre
“My charity bake sales – known as “David’s Great British Bake Off” – are always a hit, home baked products taste so much better than shop bought and are greatly appreciated by staff!”
David Jenkins
Purchasing Officer
“We’ve seen some remarkable responses, with an improvement for some patients within days. This is an early phase trial so there’s a lot more work to do. But the data we have so far is very encouraging and could help many thousands of people in the future”
Tim Somervaille
Senior Group Leader
“It is a pleasure to introduce my team who work to deliver our research goals. We work in a friendly and collaborative environment, supporting each other’s projects. “
Amaya Virós
CRUK Advanced Clinician Scientist Fellow
Our vision for world leading cancer research in the heart of Manchester
We are a leading cancer research institute within The University of Manchester, spanning the whole spectrum of cancer research – from investigating the molecular and cellular basis of cancer, to translational research and the development of therapeutics.
Our collaborations
Bringing together internationally renowned scientists and clinicians
Scientific Advisory Board
Supported by an international Scientific Advisory Board
Careers that have a lasting impact on cancer research and patient care
We are always on the lookout for talented and motivated people to join us. Whether your background is in biological or chemical sciences, mathematics or finance, computer science or logistics, use the links below to see roles across the Institute in our core facilities, operations teams, research groups, and studentships within our exceptional graduate programme.
A note from the Group Leader – Iain Hagan
We exploit genetic malleability of fission yeast as a model organism to study three aspects of the control and execution of cell division: mitotic commitment, mitotic exit and mitotic kinase function. We then apply the lessons learned from yeast to the interrogation of the same processes in the complex human cell divisions.