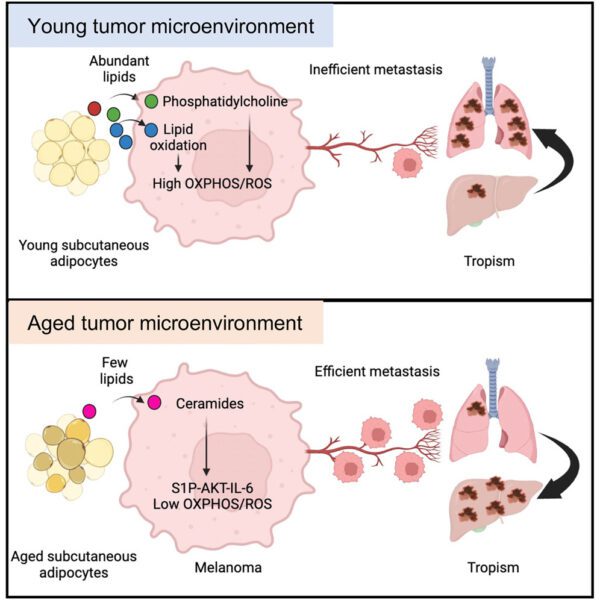
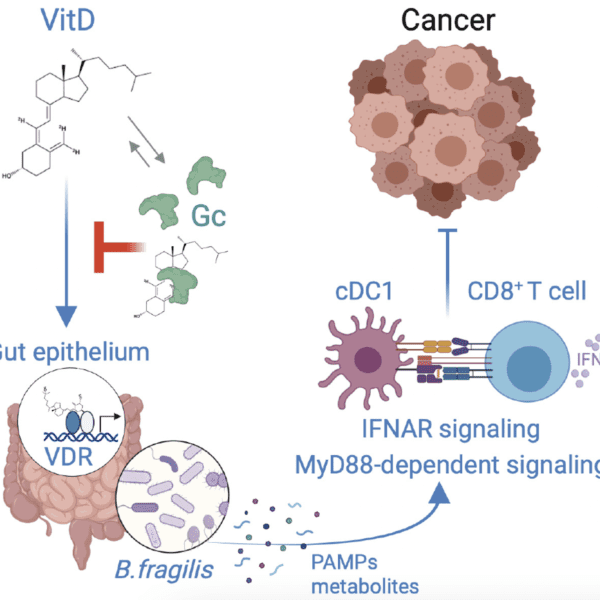
Overview
We hope to understand how somatic genetic changes and TME effects proportionally increase aggressive clonal evolution and resistance in localised prostate and penile cancer phenotypes. These factors may drive the genetic evolution in hereditary prostate cancers with BRCA2, ATM and MSH2 mutations (i.e. DDR deficient = “DDRd”) when compared to sporadic prostate cancers. Importantly, the outcomes are poor for 10-15% of high-risk localised prostate cancer patients who have these DDR germline defects. In these germline-associated DDRd mutations and prostate cancers, genetic instability is increased when compared to age-matched sporadic cancers and show behaviours that usually only occur in sporadic metastatic castrate resistant prostate cancer (mCRPC) drive new treatment options.
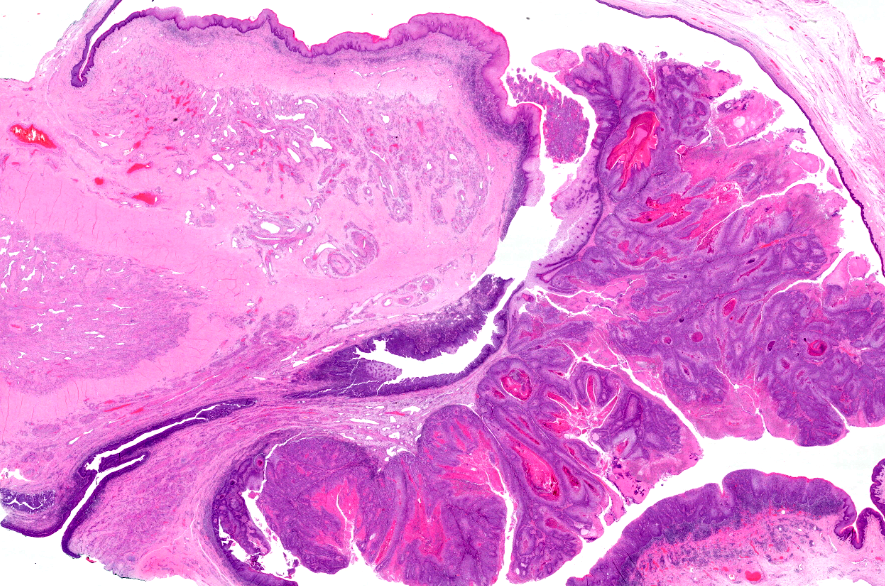
We have developed a translational pipeline to acquire normal and tumour tissues derived from gDDRd patients attending the DDR Urology clinic at the Christie NHS Foundation Trust. Patient-derived prostate epithelial cells (PrECs) have stable genomes and the ability to form 3D prostaspheres ex vivo. We also have access to Europe’s largest collection of penile cancer cases married to clinical outcome for multiomic studies of genomic and TME changes in relation to HPV subtype and differential histopathology.
In general, we are interested whether polyclonality arises from a functional loss of DDR, DNA replication fork stress and protection (c-Myc) and/or loss of cell cycle arrest (loss of TP53 or RB), and how hypoxia may modify these evolutionary processes, in both prostate and penile cancers.
Objectives:
- Evolution of Polyclonality in Cancer uses whole genome sequencing of high-risk prostate cancers and develops novel human primary epithelial cell (PrEC) isogenic cells that represent sporadic and DDRd prostate cancer models. We determine cell autonomous effects of primary and secondary somatic mutations and TME-hypoxia effects on DNA replicative stress and the absence of cell cycle checkpoints. In penile cancer, we also study HPV-subtypes and HPV programming in a similar context.
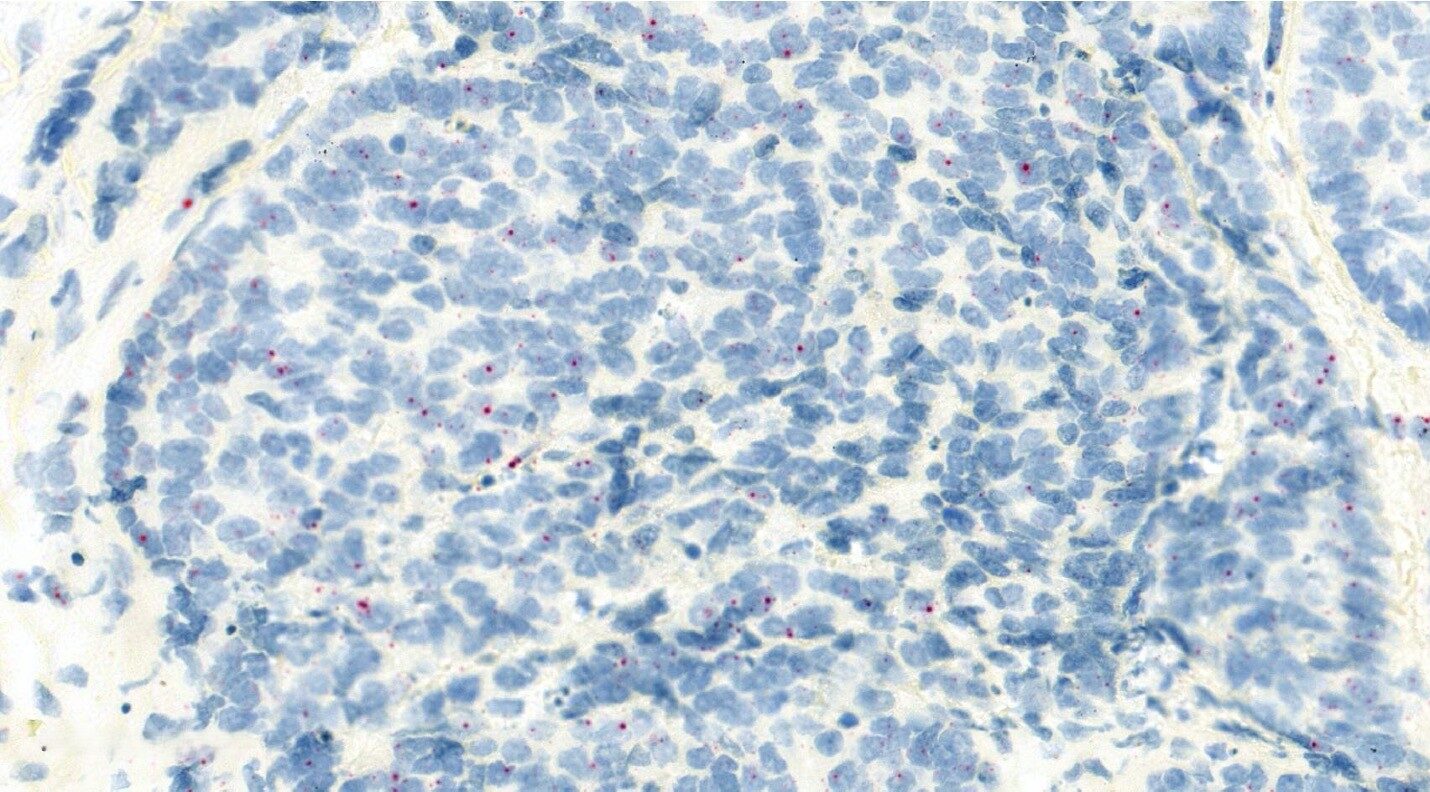
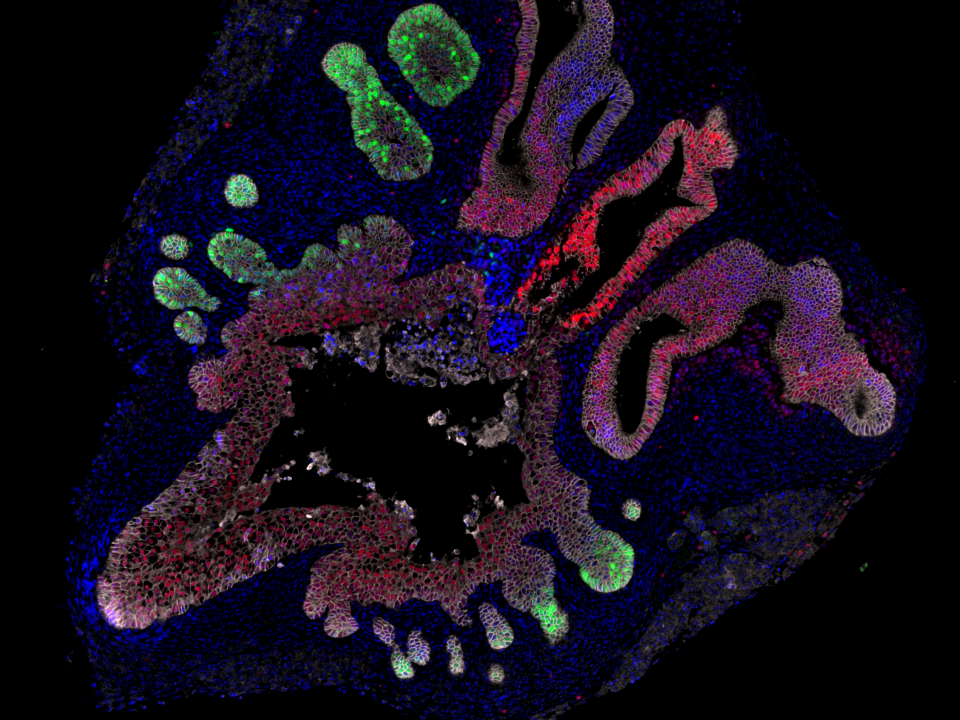
- Tumour Architecture of Aggression will define polyclonality and evotypes in hypoxic sub-regions of sporadic and hereditary prostate primary tumours and metastases and primary penile tumours using spatial multiomics relative to in situ genomic alterations. It will define polyclonality and secondary somatic mutations in situ in the context of hypoxic sub-regions, CIN/replication stress and immune cell infiltration.
- Mapping Resistance Phenotypes uses sporadic and hereditary tumour models (PrEC, DVL3, LnCaP) to test whether de novo or acquired polyclonality leads to differential Enzalutamide resistance (EnzaR) or DDR inhibitor resistance.
Featured Publications
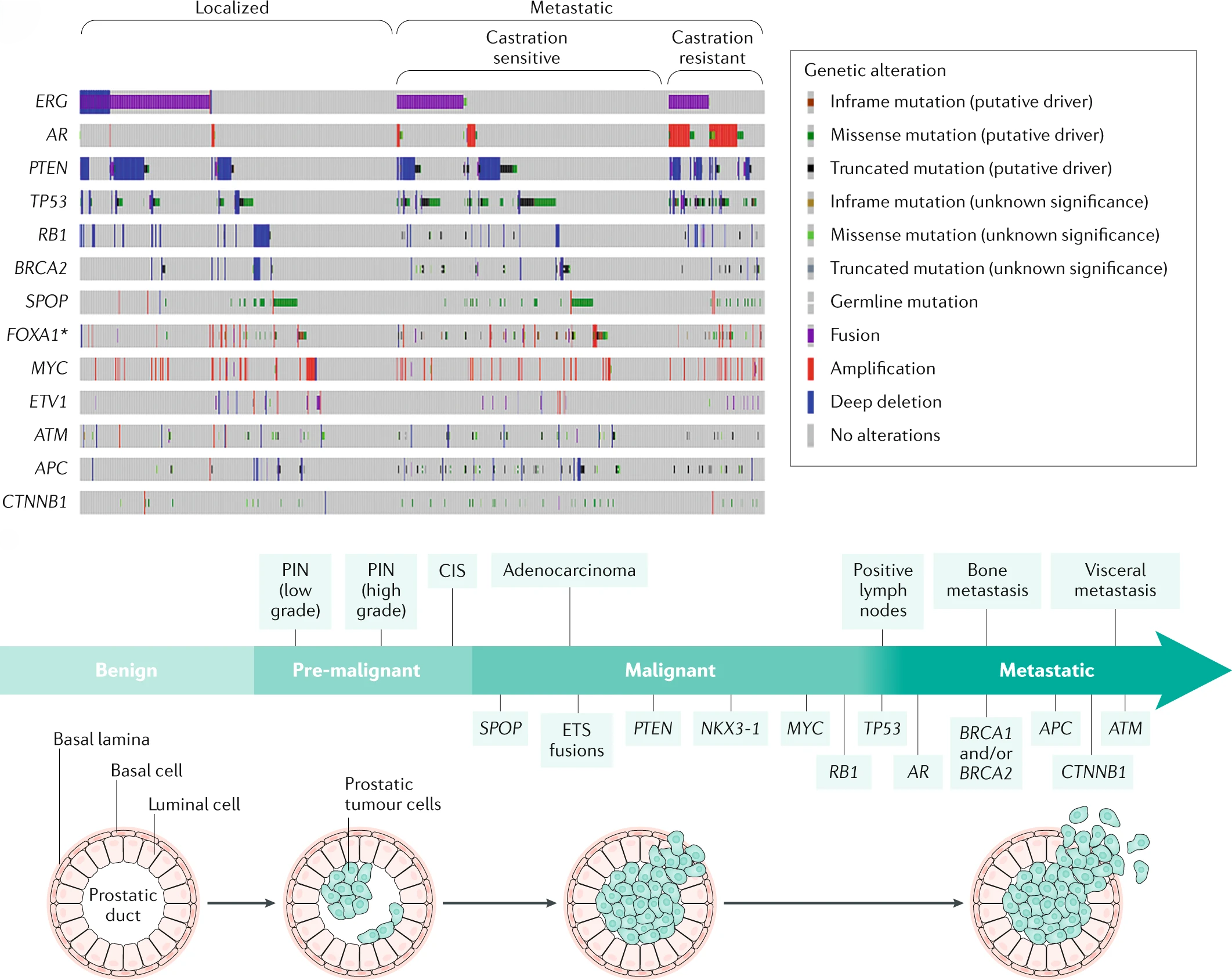
Prostate Cancer
4th February 2021
Authors discuss the epidemiology, mechanisms of disease, diagnosis, screening and prevention, management, quality of life and outlook for patients with prostate cancer.
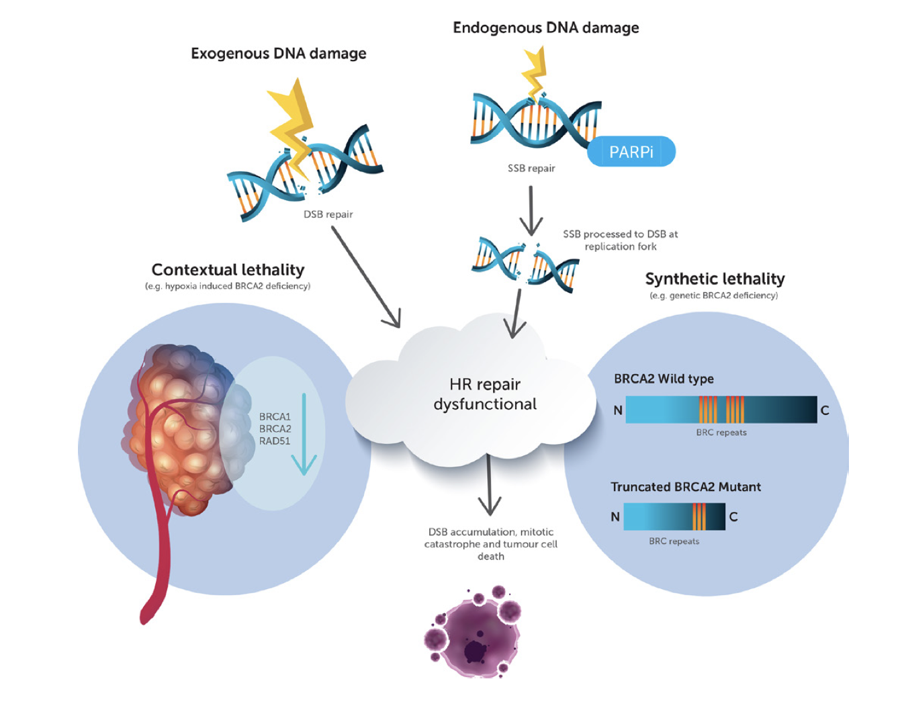
Bad neighbours: hypoxia and genomic instability in prostate cancer
1st November 2020
Authors review hypoxia and prostate tumour progression and discuss the biochemistry of hypoxia and implications for prostate cancer, targeting of hypoxic cancer cell, rational combinations for anti-hypoxia therapy and future outlook.
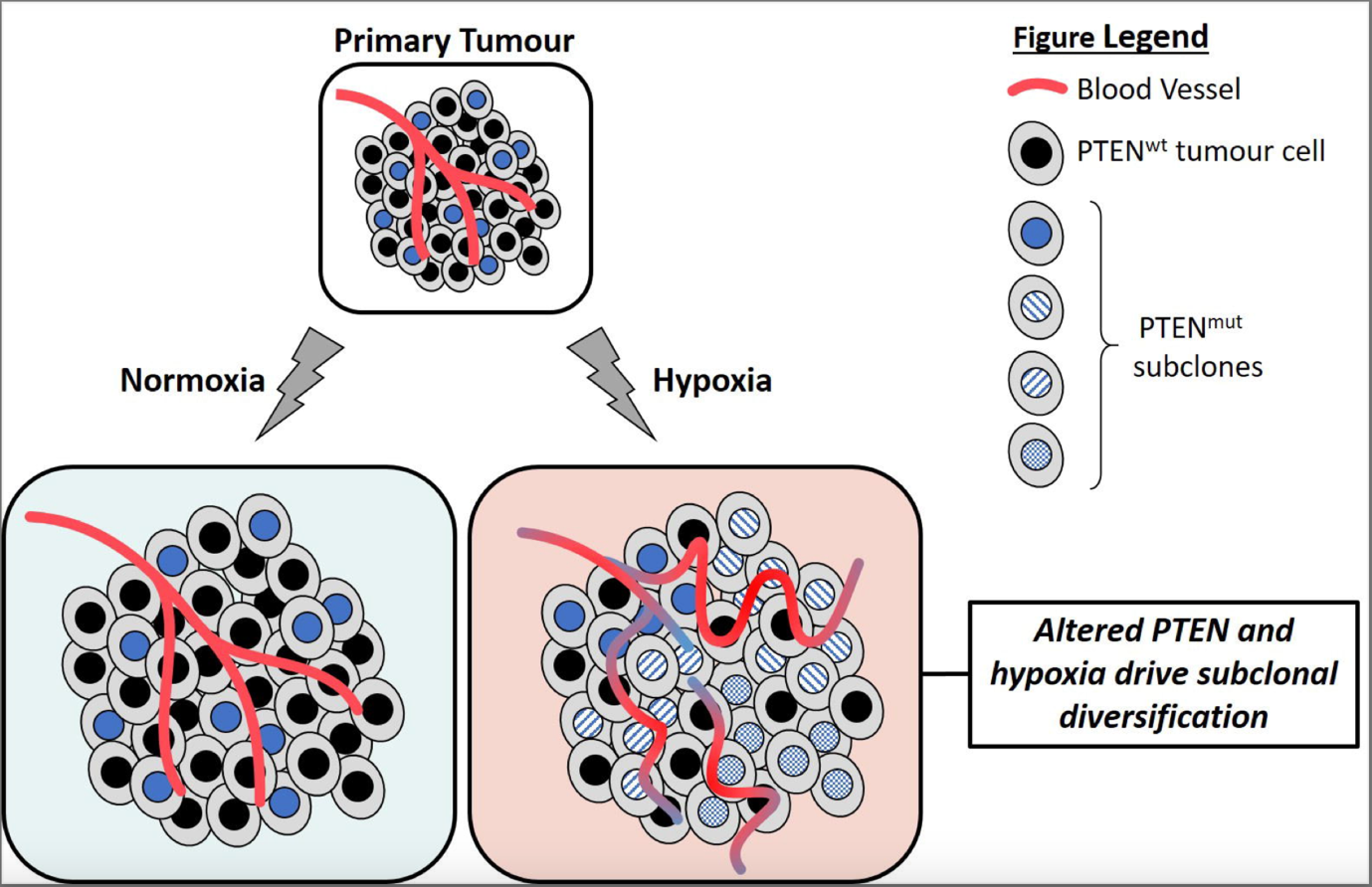
Molecular landmarks of tumour hypoxia across cancer types
14th January 2019
The authors looked at tissue hypoxia as an important differentiating metabolic characteristic between normal and malignant tissues. They show extensive intertumor- and intratumor-type heterogeneity in tumor hypoxia.
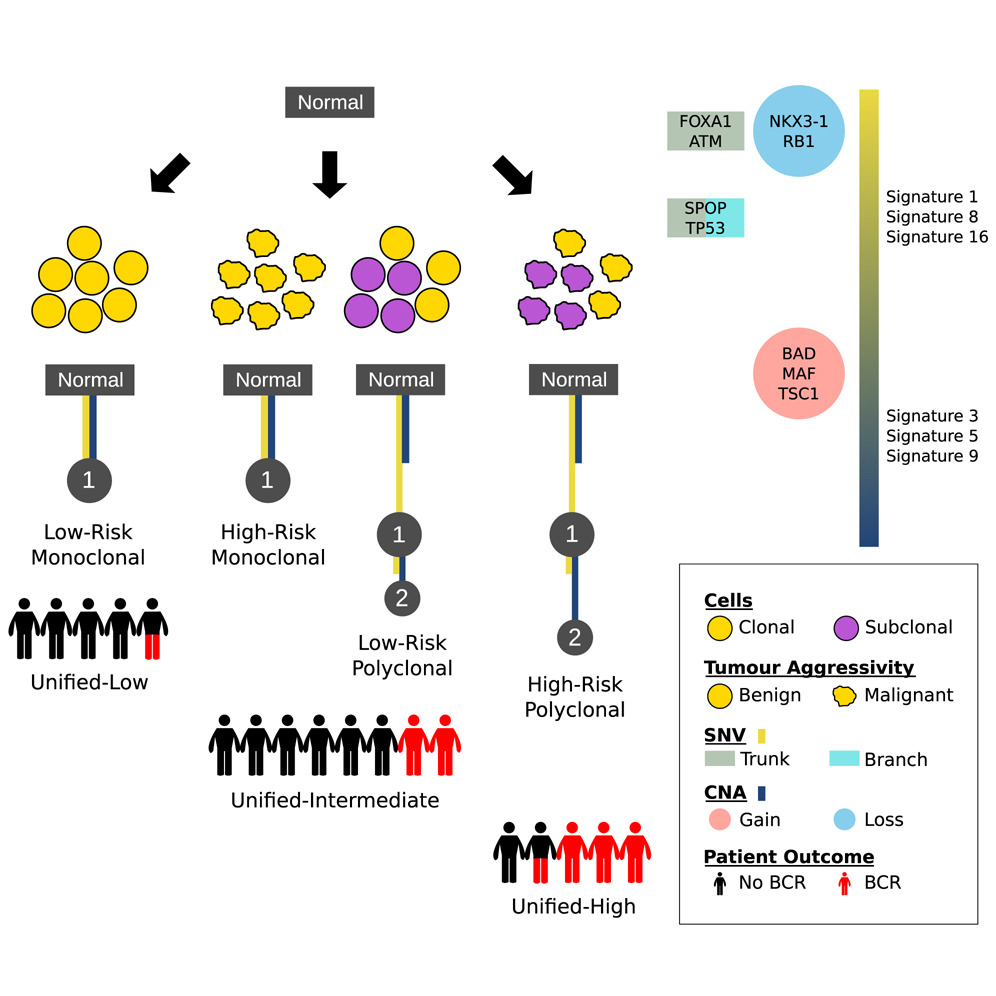
The Evolutionary Landscape of Localized Prostate Cancers Drives Clinical Aggression
19th April 2018
Multi-region sequencing reveals key aspects of tumor evolution from small numbers of patients. Authors evaluated a large cohort of localised prostate tumours to understand the population characteristics of their subclonal evolution.
Research areas
Meet the group
We have a fantastic and collegial group of scientists and clinicians working together on mechanistic and translational science within Translational Oncogenomics.


Get in touch

Institute life in Manchester
We strive to make our community a welcoming, caring and enthusiastic one, fuelling ambition with opportunities for training and mentoring to help us all achieve our personal and professional goals.

“We are so pleased to have received the funding to enable us to test our hypothesis in the lab. If we can create a new medicine that can precisely target a specific type of cell within the tumour, and restore anti-cancer immune responses, this will be a game-changer for oesophageal cancer patients “
Sara Valpione
Former Institute Clinical Fellow and now Clinician in Residence within the CRUK National Biomarker Centre

“My charity bake sales – known as “David’s Great British Bake Off” – are always a hit, home baked products taste so much better than shop bought and are greatly appreciated by staff!”
David Jenkins
Purchasing Officer

“We’ve seen some remarkable responses, with an improvement for some patients within days. This is an early phase trial so there’s a lot more work to do. But the data we have so far is very encouraging and could help many thousands of people in the future”
Tim Somervaille
Senior Group Leader

“It is a pleasure to introduce my team who work to deliver our research goals. We work in a friendly and collaborative environment, supporting each other’s projects. “
Amaya Virós
CRUK Advanced Clinician Scientist Fellow
“We are so pleased to have received the funding to enable us to test our hypothesis in the lab. If we can create a new medicine that can precisely target a specific type of cell within the tumour, and restore anti-cancer immune responses, this will be a game-changer for oesophageal cancer patients “
Sara Valpione
Former Institute Clinical Fellow and now Clinician in Residence within the CRUK National Biomarker Centre
“My charity bake sales – known as “David’s Great British Bake Off” – are always a hit, home baked products taste so much better than shop bought and are greatly appreciated by staff!”
David Jenkins
Purchasing Officer
“We’ve seen some remarkable responses, with an improvement for some patients within days. This is an early phase trial so there’s a lot more work to do. But the data we have so far is very encouraging and could help many thousands of people in the future”
Tim Somervaille
Senior Group Leader
“It is a pleasure to introduce my team who work to deliver our research goals. We work in a friendly and collaborative environment, supporting each other’s projects. “
Amaya Virós
CRUK Advanced Clinician Scientist Fellow
Our vision for world leading cancer research in the heart of Manchester
We are a leading cancer research institute within The University of Manchester, spanning the whole spectrum of cancer research – from investigating the molecular and cellular basis of cancer, to translational research and the development of therapeutics.
Our collaborations
Bringing together internationally renowned scientists and clinicians
Scientific Advisory Board
Supported by an international Scientific Advisory Board
Careers that have a lasting impact on cancer research and patient care
We are always on the lookout for talented and motivated people to join us. Whether your background is in biological or chemical sciences, mathematics or finance, computer science or logistics, use the links below to see roles across the Institute in our core facilities, operations teams, research groups, and studentships within our exceptional graduate programme.
A note from the Group Leader – Rob Bristow
A cancer evolves over time and becomes aggressive secondary to increasing genomic instability and a permissive tumour microenvironment (TME) based on decreased immune surveillance and metabolic changes including intratumour hypoxia. We believe that both sporadic and hereditary prostate cancers increase their metastatic potential by increasing somatic mutations on the background of germline SNPs or mutations in genes involving DNA damage response and repair. We are also studying the genetic and TME factors that determine relative aggression in rare penile cancers given Manchester is an internationally renowned clinical centre for the treatment of this disease.
My laboratory is a translational one which practices cancer team science whereby scientists and clinicians work together to answer some of cancer’s most important issues with multiomic technologies including spatial transcriptomics and proteomics. We use primary human tumour samples coming directly from the clinic and are developing model systems in prostate cancer based on these clinical specimens to create patient-faithful cell lines and xenografts in hereditary and sporadic prostate cancer. These models can also be characterised for their response to experimental radiotherapy and/or targeted therapies such as PARP inhibitors based on genetic mutations-based or TME-based synthetic lethality.