
Professor Iain Hagan
Cell Division Group Leader
Iain is a Senior Group Leader at the Institute, running the Cell Division lab. He is passionate about the study of cell division and cell cycle control. Using sophisticated manipulation of yeast as a model system, he has defined fundamental principles of cell cycle control and division that have informed our understanding of cancer cell proliferation to refine therapeutic approaches.
About Professor Iain Hagan
After completing his PhD studies Iain Hagan went to Japan on a 4-year postdoctoral fellowship with Professor Mitsuhiro Yanagida in Kyoto University. He returned to the UK in 1993 with a Cancer Research Campaign Return Fellowship to establish a group in The University of Manchester. He continued to work in what later became the Faculty of Life Sciences at The University of Manchester, with further Cancer Research Campaign Fellowship support before moving to the Cancer Research UK Manchester Institute in 2001.
In 1999, Iain was awarded the Human Frontier Science Program 10th Anniversary Medal and in 2001 was the recipient of the BSCB Hooke Medal. Iain is currently a Senior Group Leader at the CRUK Manchester Institute. In 2009, he was elected to full membership of the European Molecular Biology Organisation (EMBO). In 2016, Iain was awarded a Wellcome Trust Investigator Award to study spatial and temporal control of mitotic commitment.
Iain is Chair of the internal Grants Committee that reviews all our grant applications. From 2021 until 2023, he was Deputy Director at the CRUK Manchester Institute.
Groups
Qualifications
- PhD in Cell Biology | 1987 | University College London & ICRF
- BSc in Biochemistry | 1984 | University College London
Interests
- Control and execution of mitosis
- Control of protein phosphatases
- Mitotic spindle
Research Projects
Publications
- PKMYT1 has an important role in the timing and fidelity of chromosome segregation
- Release from cell cycle arrest with Cdk4/6 inhibitors generates highly synchronized cell cycle progression in human cell culture
- Elevated basal AMP-activated protein kinase activity sensitizes colorectal cancer cells to growth inhibition by metformin
- A TOR (target of rapamycin) and nutritional phosphoproteome of fission yeast reveals novel targets in networks conserved in humans
Why I work at CRUK MI
“The Institute is made of some fantastic people who are dedicated to their research and want to make a difference to the lives of cancer patients. It is a real pleasure and honour to work alongside these great scientists and to nurture our early career researchers.”
Visit Research Group
The inappropriate proliferation of cancer cells can arise from unchecked cell division, a failure to engage cell death pathways, or simultaneous changes in both. Understanding how the diverse cues are integrated to co-ordinate cell division and death is therefore key to understanding the biology of cancer. The Cell Division group study cell cycle controls that determine when a cell commits to the physical process of genome segregation, mitosis.
Because the regulatory networks that control cell division are highly conserved, we use both unicellular fission yeast and human cells in our investigations as the yeast work identifies core principles with which to frame questions to ask of the more complex context of human cell division. Our yeast work addresses how signals from the broad range of pathways are integrated by regulatory relays on neighbouring scaffolds on the centrosome to generate a single signal to trigger division when the time is right.
Complementary studies are asking whether similar controls operate in human cells and characterises one of the key cell cycle checkpoint molecules that determines when mitosis begins, PKMYT1.
Get in touch
https://www.cell.com/cell/fulltext/S0092-8674(26)00522-2
Plasma signals of lung tumor promotion for molecular cancer prevention
25 June 2026
Institute Authors (1)
William Hill
Research Group
Cancer Origins
25 June 2026
https://link.springer.com/article/10.1038/s44319-026-00809-1
PKMYT1 has an important role in the timing and fidelity of chromosome segregation
5 June 2026
Institute Authors (5)
Asma Belbelazi, Charlie Greenaway-Wells, Zoe Edwards, Keren Dawson, Iain Hagan
Research Group
Cell Division
5 June 2026
https://www.nature.com/articles/s42255-026-01514-y
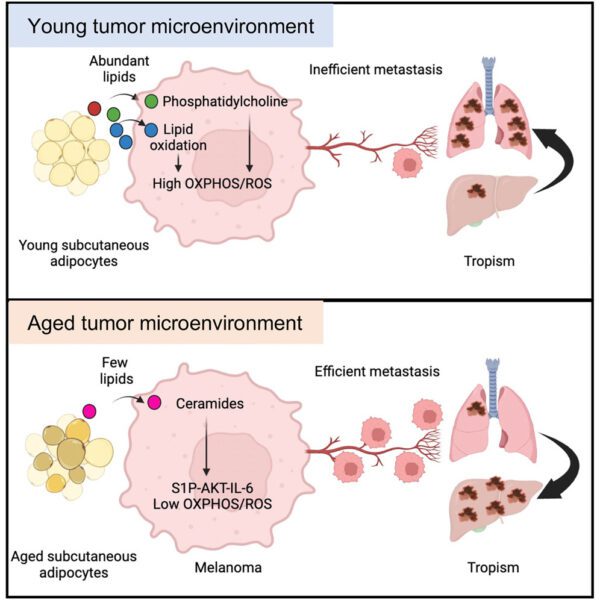
Tissue-specific fibroblast lipid cues impose the rate of epithelial cancer invasion
27 April 2026
Institute Authors (4)
Amaya Virós, Noah Palombo, Charlotte Russell, Claus Jørgensen
Research Group
Skin Cancer & Ageing
27 April 2026
https://www.cell.com/cancer-cell/fulltext/S1535-6108(26)00114-5
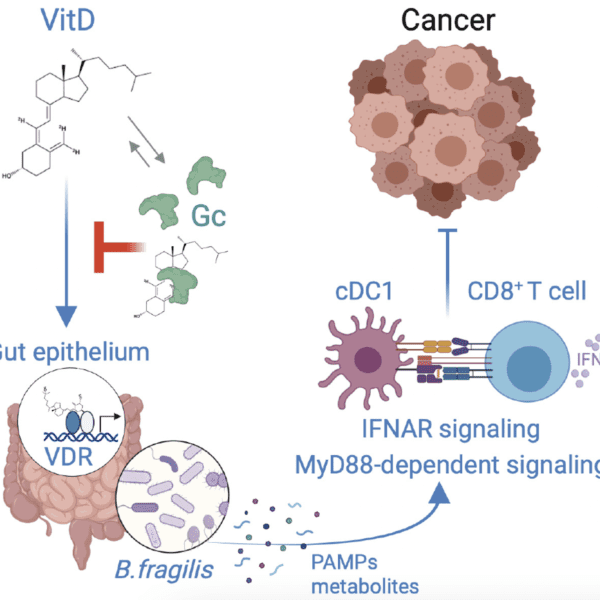
Immunometabolic gatekeeping: How tissue metabolism conditions tumor immunity
13 April 2026
Institute Authors (1)
Samra Turajlić
Research Group
Cancer Dynamics
13 April 2026
https://www.nature.com/articles/s41467-026-69964-2
Disruption of tRNA threonylation triggers RIG-I mediated anti-tumour immune response
25 February 2026
Institute Authors (1)
Sylvain Delaunay
Research Group
RNA Dynamics in Cancer
25 February 2026
https://doi.org/10.1038/s44161-025-00740-z
Single-cell profiling reveals three endothelial-to-hematopoietic transitions with divergent isoform expression landscapes
11 November 2025
Institute Authors (6)
Robert Sellers, John Weightman, Wolfgang Breitwieser, Natalia Moncaut, Michael Lie-a-ling, Georges Lacaud
Labs & Facilities
Computational Biology Support, Molecular Biology, Genome Editing and Mouse Models
Research Group
Stem Cell Biology
11 November 2025
Our vision for world leading cancer research in the heart of Manchester
We are a leading cancer research institute within The University of Manchester, spanning the whole spectrum of cancer research – from investigating the molecular and cellular basis of cancer, to translational research and the development of therapeutics.
Our collaborations
Bringing together internationally renowned scientists and clinicians
Scientific Advisory Board
Supported by an international Scientific Advisory Board
Careers that have a lasting impact on cancer research and patient care
We are always on the lookout for talented and motivated people to join us. Whether your background is in biological or chemical sciences, mathematics or finance, computer science or logistics, use the links below to see roles across the Institute in our core facilities, operations teams, research groups, and studentships within our exceptional graduate programme.

Institute life in Manchester
We strive to make our community a welcoming, caring and enthusiastic one, fuelling ambition with opportunities for training and mentoring to help us all achieve our personal and professional goals.

“We are so pleased to have received the funding to enable us to test our hypothesis in the lab. If we can create a new medicine that can precisely target a specific type of cell within the tumour, and restore anti-cancer immune responses, this will be a game-changer for oesophageal cancer patients “
Sara Valpione
Former Institute Clinical Fellow and now Clinician in Residence within the CRUK National Biomarker Centre

“My charity bake sales – known as “David’s Great British Bake Off” – are always a hit, home baked products taste so much better than shop bought and are greatly appreciated by staff!”
David Jenkins
Purchasing Officer

“We’ve seen some remarkable responses, with an improvement for some patients within days. This is an early phase trial so there’s a lot more work to do. But the data we have so far is very encouraging and could help many thousands of people in the future”
Tim Somervaille
Senior Group Leader

“It is a pleasure to introduce my team who work to deliver our research goals. We work in a friendly and collaborative environment, supporting each other’s projects. “
Amaya Virós
CRUK Advanced Clinician Scientist Fellow
“We are so pleased to have received the funding to enable us to test our hypothesis in the lab. If we can create a new medicine that can precisely target a specific type of cell within the tumour, and restore anti-cancer immune responses, this will be a game-changer for oesophageal cancer patients “
Sara Valpione
Former Institute Clinical Fellow and now Clinician in Residence within the CRUK National Biomarker Centre
“My charity bake sales – known as “David’s Great British Bake Off” – are always a hit, home baked products taste so much better than shop bought and are greatly appreciated by staff!”
David Jenkins
Purchasing Officer
“We’ve seen some remarkable responses, with an improvement for some patients within days. This is an early phase trial so there’s a lot more work to do. But the data we have so far is very encouraging and could help many thousands of people in the future”
Tim Somervaille
Senior Group Leader
“It is a pleasure to introduce my team who work to deliver our research goals. We work in a friendly and collaborative environment, supporting each other’s projects. “
Amaya Virós
CRUK Advanced Clinician Scientist Fellow